在加载过程中,弹簧所吸收的能量称为变形能,以U 表示。对于自由放置的螺旋弹簧,在变形恢复时若没有与其他零件相摩擦,摩擦损失的能量等于零,则弹簧放出的能量和积蓄的能量相等。对于叠板弹簧、碟形弹簧及环形弹簧等,由于在过程中弹簧间变形相互摩擦,故有相当数量的摩擦功存在,致使卸载时的特性线低于加载时的特性线,如图3-35 所示。设计缓冲弹簧时,为保证其缓冲能力,应要求弹簧的变形能大于 被缓冲物体的动能。......
2025-09-29
1.钝化的特征与途径
由于阳极过程受到阻滞而引起金属或合金耐蚀性提高的现象称为钝化。金属钝化后所处的状态称为钝态。钝态金属所具有的性质称为钝性。金属钝化后形成的钝化膜(可以是成相膜,也可以是吸附膜)主要是阻碍金属离子的溶解反应,也就是阻碍离子导电性,因此对阳极极化的超电压影响很大,使电位向正方向强烈偏移。钝化膜对起因于电子导电性受阻的阴极极化的影响要小得多。钝化后的主要特征:首先金属的电极电位向正方向强烈偏移,如Fe钝化后,电极电位可以从-0.5~0.2V升高到0.5~1.0V;Cr钝化后,电极电位从-0.6~-0.4V升高到0.8~1.0V;其次是腐蚀速度急剧降低,钝化的腐蚀速度有时可减小到正常腐蚀速度的数百分之一到数千分之一。
使金属钝化的方法有两种,一种是利用氧化剂的化学钝化法,钢铁的发蓝就是用NaNO2和NaOH在高温下使金属表面生成一层蓝黑色的钝化膜。如果金属能被空气中的氧气钝化,该金属就称为自钝化金属,如铝、钛等。当自钝化金属表面的钝化膜遭到破坏时,其往往具有自愈能力,因此具有很好的耐蚀性。另一种方法是用外加电源的阳极钝化法,如18-8不锈钢在30%H2SO4中会剧烈地腐蚀,但如果通以一定的阳极电流,就能使其腐蚀速度迅速下降到原来的数万分之一,这种现象就称为阳极钝化或电化学钝化。
典型的具有钝化特征的金属电极的阳极极化曲线如图1-21所示。图中φe是没有外加阳极极化时的金属平衡电极电位,从φe到φp(钝化电位)之间是金属电极的活性溶解区,当阳极极化电位等于φp时,金属阳极溶解电流密度达到最大值,称为致钝电流密度im。从φp到φcp(完全钝化电位,维钝电位)是活化-钝化过渡区,此时金属由活性态转变为钝态,阳极电流密度急剧下降。
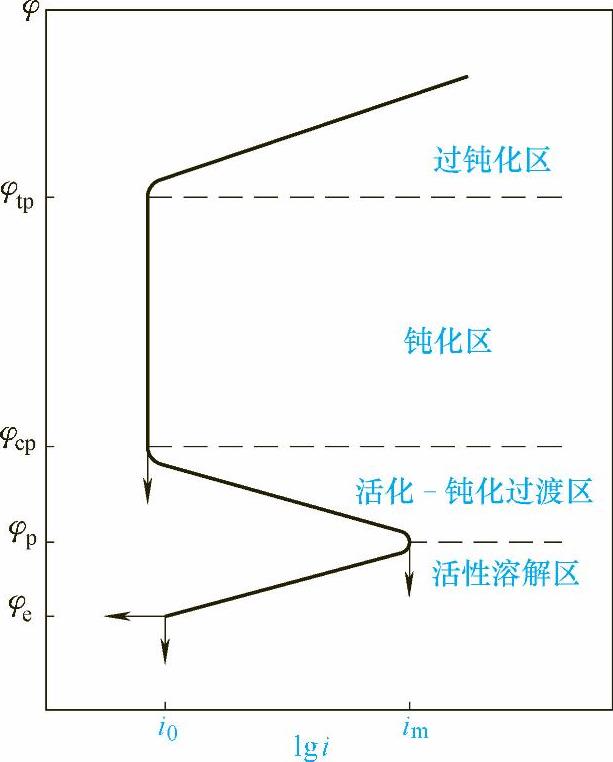
图1-21 金属钝化过程的阳极极化曲线示意图
在活化-钝化过渡区,金属表面处于不稳定状态,从φp到φcp的电位区间内,有时电流密度会出现剧烈振荡。从φcp到φtp(过钝化电位)之间是钝化区,金属表面处于稳定的钝化状态,金属的阳极溶解电流密度很低,并且基本上不随电位而变化。当电位高于φtp时进入过钝化区,阳极电流密度再次随电位的升高而增大。电流重新增大的原因,一种可能是电极上发生了新的反应,如析氧反应;另一种可能是钝化膜进一步氧化而生成高价的易溶化合物,因而加速了腐蚀的进行。
上面所讲的是理想的阳极极化曲线,实际上在阳极极化的同时必然存在一个共轭的去极化剂的阴极极化过程。所以实测的阳极极化曲线是由金属的真实阳极极化曲线与阴极极化曲线合成而得的表观阳极极化曲线。表观阳极极化曲线所对应的外加阳极电流等于真实阳极电流与去极化剂的阴极反应电流之差。因此,金属在介质中是否处于钝态,不仅取决于真实阳极极化曲线钝化区范围的大小,同时也取决于去极化剂阴极极化曲线的相对位置。对于实测的阳极极化曲线在此不加以详细阐述。
2.钝化理论
尽管钝化现象已研究了100多年,积累了大量的表面现象,但对其研究范畴、作用机理等仍然存在着分歧,甚至对金属钝化的确切定义也还没有取得一致的意见,因此只能对目前并存的、主要的两种解释金属钝化现象的学说——成相膜和吸附理论进行比较和介绍。
(1)成相膜理论 成相膜理论又称薄膜理论,该理论认为,当金属溶解时,可在表面上生成致密的、覆盖性良好的保护膜,这种保护膜作为一个独立的相而存在,并把金属与溶液机械地隔离开,使金属的溶解速度大大降低,即使金属转为钝态。
支持成相膜理论的实施证据主要有:
1)用I2-KI溶液作溶剂可溶解基体金属而分离出Fe的钝化膜。
2)用椭圆偏振仪可测得钝化膜的厚度,Fe膜厚度为25~30Å,碳钢膜厚度为90~110Å,不锈钢膜为9~10Å。
3)用电子衍射法对钝化膜进行相分析证明,大多数的钝化膜由金属氧化物组成,如Fe的钝化膜是γ-Fe2O3,Al的钝化膜下层为无孔的γ-Al2O3,上层为多孔的β-Al2O3·3H2O。这些事实都证明了成相膜理论。
(2)吸附理论 吸附理论认为引起金属钝化并不一定要形成成相膜,而只要在金属表面或部分表面上生成氧或含氧粒子的吸附层就足够了。这一吸附层至多只是单分子层(二维的),它可以是OH-或O2-离子,但更多人认为可能是氧原子。氧原子和金属的最外层原子因化学吸附而结合,使金属表面的化学结合力饱和,并改变了金属/溶液界面的结构,大大提高了阳极反应的活化能,故金属与腐蚀介质的化学反应将显著减小,这就是钝化的原因。所以支持吸附理论者认为,出现钝化是由于金属本身反应能力的降低,而不是由于膜的机械隔离作用。
支持吸附理论的实验证据主要有:
1)Pt在盐酸中,当它的6%表面积被吸附氧覆盖时,其电位朝正方向移动0.12V,Pt的溶解速度降为原来的1/10,当充满12%的表面积时,溶解速度降为1/16。
2)Fe在0.05mol/L的NaOH溶液中用1×10-5A/cm2恒电流极化时,只需通过0.3mq/cm2的电量就可使Fe电极钝化,但这些电量不足以形成成相膜。
3)已知在某些条件下,电极电位可在相应于活化态与钝态之间振荡,这也说明钝态的建立和破坏只需要少量的电量转移就能实现。
4)Uhlig发现不论用何种方法得到的Fe的钝化膜,其吸附氧的化学当量实验值都约为0.01q/cm2,这一数量相当于铁表面形成一个氧原子层(8×1014个吸附氧原子/cm2)加单分子层氧分子(2×1015个氧分子/cm2)的化学吸附层,因而提出Fe的吸附钝化膜可用O2·O·Fe来表示。
5)金属钝化的难易取决于金属对氧吸附能力的大小,以Fe和Cr的比较为例
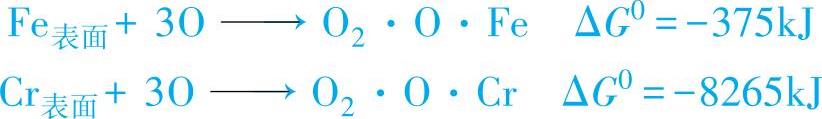
由ΔG0值可见,Cr表面对氧的吸附明显比Fe对氧的吸附稳定,可见Cr比Fe更容易钝化。
以上实验说明,金属表面的单分子吸附层甚至可以是不连续的,不一定需要将表面完全覆盖。可以设想,只要在最活泼的最先溶解的表面区域上吸附了单分子层,便能抑制阳极过程,使金属钝化。这种最活泼、最先溶解的表面区域在化学上称为活化中心,就是金属学上所指的晶格缺陷、晶格畸变、位错露头、晶粒边界等处。
吸附键形成的原因可用电子排布理论来解释。可钝化金属主要是过渡金属Fe、Ni、Cr等,由于过渡金属原子具有未充满的d电子层,它们和带有孤对电子的氧很容易形成强的化学键结合,导致氧在这些金属表面的化学吸附。从热力学上讲,过渡金属表面的氧吸附层比氧化物更稳定,因而显示钝化特性。但非过渡金属如Zn、Cd、Cu、Pb等,在室温下却是形成氧化物比保持吸附结构更为有利,因而不显示钝化特性。
(3)吸附理论和成相膜理论的统一 两种理论都能解释一部分实验现象,那么哪一种理论更为正确呢?支持吸附理论者认为,某些金属表面上只要通过不足以形成单原子氧化层的电量就可以使金属钝化。但支持成相膜理论者认为,很难证明极化前电极表面上确实完全不存在氧化膜,因此就难以判断所通过的电量是用来“建立”氧化膜还是“修补”氧化膜。如果是“修补”氧化膜,需要的电量显然要少得多。
其实两种理论并无原则性差别。首先从作用来看,成相膜和吸附膜只有厚度的差别,而其阻化作用是完全相同的。其次,成相膜的氧化物膜,其形成键是化学键;吸附膜的吸附氧层,其形成键是化学吸附键。当电极上的电极电位很正时,各阴离子与电极表面间的吸附键也逐渐具有化学键的性质,故两种理论的差别并不像表面上那么大。
一些研究者认为,金属钝化的难易主要取决于对氧吸附能力的大小(吸附理论);而能否维持钝态,却取决于钝化结果所形成的钝化膜(成相膜理论)。所以可以这样认为,金属的钝化首先是在金属的表面上形成吸附层(含氧粒子参加电化学反应后直接形成的第一层氧化层),使金属的溶解速度大幅度下降,然后在这层氧化层的基础上继续生长,形成成相膜后就具有较强的保持钝态的能力,因而绝大多数对金属溶解具有实际保护价值的钝化膜,可认为均与成相膜有关。
3.Flade(弗拉德)电位
(1)Flade电位的定义 被阳极电流钝化的金属,若把阳极电流中断,则金属的钝态很快消退而回到活化状态,测量此过程的电位-时间曲线,观察阳极电流中断后电极电位的变化,发现电位开始很快下降,然后有一段电位缓慢下降的阶段(约几秒到几分钟),最后电位又很快下降,此时金属回到活化状态,如图1-22所示。金属刚好回到活化状态的这个电位称为Flade电位φF。
Flade电位是钝态时的特性电位,首先是F.Flade在H2SO4溶液中研究钝态Fe自然活化时,在电位衰减曲线上所看到的停滞电位,后来K.F.Bonhoeffer等从活化态向钝化态转变时也测得了这一特性电位。因此,Flade电位是活化态和钝态之间的临界电位。
(2)φF与pH值的关系 φF随金属的种类和溶液pH值的变化,可表示为
φF=φ0F-0.059pH (1-48)
φ0F是pH=0时的Flade电位,称为标准Flade电位。各种金属在25℃时的标准Flade电位(SHE)为(https://www.chuimin.cn)
Fe:φ0F=0.63V
Ni:φ0F=0.22V
Cr:φ0F=-0.22V
Cr(25%)-Fe合金:φ0F=-0.10V
φF与pH值的关系:一般pH值越大,电位越负移,金属越易钝化。
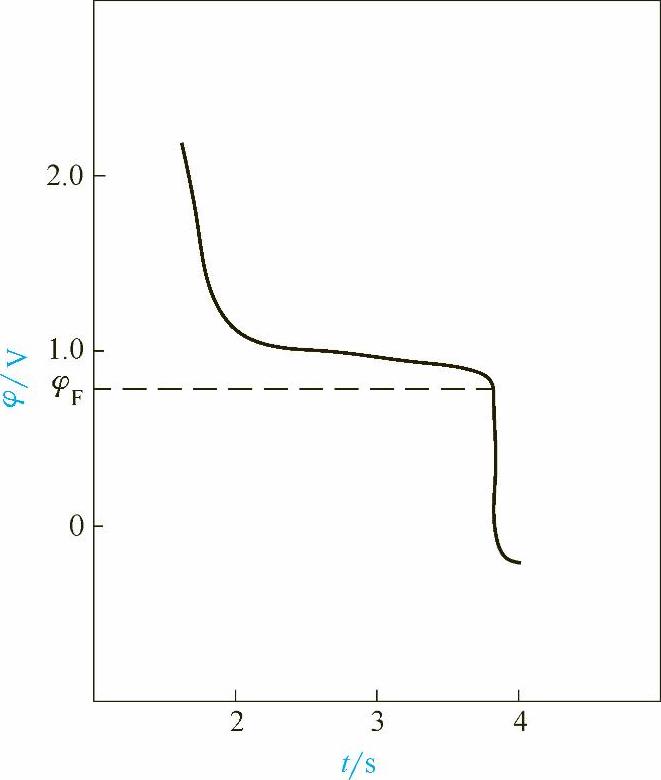
图1-22 Fe在1mol/L的H2SO4中钝态的消退情况
(3)φF的意义
1)φF可用来衡量金属钝态的稳定性。Flade电位与钝化膜的稳定性有关,是关联着钝化的生成和破坏的电位。φF越负,生成的钝化膜越稳定。
对同种金属来说,pH值越小,φF越大,表示钝化膜具有明显的活化倾向。不同金属在同样的介质中,φF下降,钝化膜的稳定性增加,对Cr-Fe合金来说,随着含Cr量的增加,φ0F从纯Fe的0.63V逐渐减小,特别在Cr质量分数为10%~15%的范围内变化更快,当Cr质量分数达到25%时,φ0F约为-0.1V。因此,Cr比Fe易钝化,Fe-Cr合金比纯Fe易钝化,而且含Cr量越多越易钝化。
2)φF对应于钝态氧化物形成反应的平衡电位。如果金属在阳极钝化过程中进行如下反应:
Me+H2O→MeO+2H++2e
对此反应,φF相当于此反应的氧化电位,而MeO是金属上的钝化膜。若设想和金属结合的氧量不会影响到所给的公式,则得
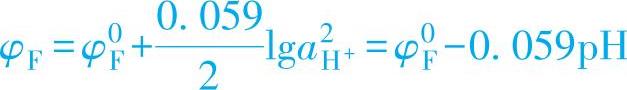
3)φF对应于金属表面的氧化物组成由低价转化为高价的平衡电位。如果活化-钝化转变是通过金属表面的氧化物组成由低价转化为高价而进入钝态的,那么Flade电位即为该转变反应的平衡电位。Vetter认为Fe表面上的钝化膜是Fe/Fe3O4/γ-Fe2O3/H2O,故Flade电位相当于下列反应的平衡电位:
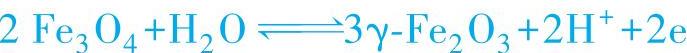
Nagayama认为中性溶液中Fe的Flade电位和[Fe2+]有关,提出Flade电位相应于下列反应的平衡电位:

一般从热力学数据求得的计算值与实测值相差很大(约0.6V),K.J.Vetter认为这差别是由于钝化膜内Fe原子的化学位不同所产生的电位差,即表面自由能变化的影响之故。
4)φF对应于介稳态氧化物相形成的最低电位。如果金属的钝化是由于形成了介稳态的氧化物相,那么Flade电位与介稳态氧化物相形成的最低电位相对应。大多数金属在酸性溶液中,按照热力学计算似乎不可能生成固相产物,然而却出现了金属的钝化现象,此即因为生成了介稳态化合物。介稳态通常在氧化膜的生成速度大于氧化膜的溶解速度时生成。
5)Flade电位φF与致钝电位φp很接近,但并不相同。首先,φF是钝化膜活化的电位,而φp是产生钝化的电位。其次,φF是在断电后测得的,而φp是在通电状态下测得的,故包含有钝化膜的电阻降和膜孔隙中溶液的浓差极化。因此两者之间可相差几十毫伏,有时也可以把φp近似地看作φF。但也有人认为φF应该与φcp(维钝电位)更为类似,因为它是由钝态变到活化态的特征电位。
4.过钝化与钝态被破坏引起的腐蚀
(1)过钝化与二次钝化 金属随着电位的增加,从钝态重新转变为活性溶解的过程称为过钝化,其主要原因是金属在强烈的阳极极化或强氧化剂的作用下形成了可溶性的或不稳定的高价化合物。处于过钝态的金属具有相当大的腐蚀电流,但过钝化所引起的腐蚀是均匀腐蚀。
过钝化相当于二次活化,当过钝化后电位继续正移时,又可能出现二次钝化。例如不锈钢在阳极极化过程中,由于Cr在过钝化区的选择性溶解,使Fe在金属表面上的含量增加,因为Fe的钝化区间比Cr宽,因而造成二次钝化。
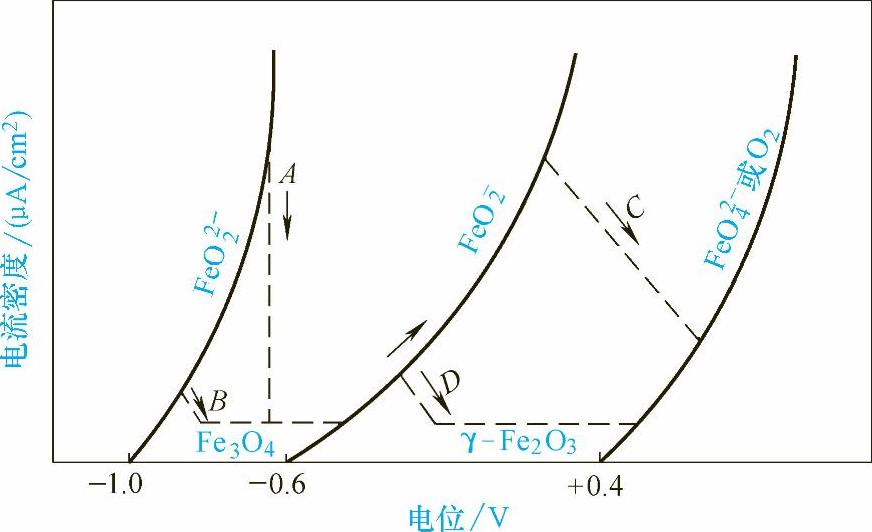
图1-23 Fe在140℃的48%NaOH溶液中的阳极极化行为
图1-23是Fe在140℃的48%NaOH溶液中的阳极极化行为。当电位升高时,Fe首先以FeO22-离子溶出,然后在-0.9~-0.8V时钝化,极化速度较慢时,极化曲线从高电流密度的A点移向钝化区,极化速度较快时,从低电流密度的B点移向钝化区,此时金属表面上生成Fe3O4膜。电位继续升高时,金属呈FeO-2离子溶出。当电位再次升高时,就会发生二次钝化,D线就是因为生成γ-Fe2O3而进入稳定钝化区,钝化环境保持到FeO24-生成或析氧反应发生。
(2)氯离子对钝化的影响 Br-、I-离子具有与Cl-离子相似的作用。空气中钝化的铬在H2SO4溶液中可保持钝化状态,但若在H2SO4溶液中添加NaCl,经过一定时间后铬就开始腐蚀并有H2放出,这就是Cl-离子对钝化膜的破坏作用。
1)成相膜理论的解释。为了说明Cl-离子破坏钝化膜而产生点蚀的原因,Hoar从成相膜理论出发提出了离子交换理论。由于Cl-离子易于变形和极化,当它在氧化膜表面吸附时在膜上产生了一个很强的诱导电场,此电场能将金属阳离子拉出氧化物晶格而进入溶液相;也能使Cl-离子向氧化物晶格内部渗透而发生离子交换作用。这两个作用都使氧化物晶格产生阳离子空位,增加了氧化物的离子导电性,使阳离子更易在电场作用下迁移,最终导致小孔腐蚀。因此氧化膜中产生足够强的电场是钝化膜破坏的关键。诱导电场的强度取决于Cl-离子浓度和电极电位,故膜的破坏需要一个最低的Cl-离子浓度(临界浓度)和一个最低的电位(孔蚀击破电位),它的数值与电解质溶液的组成有关。Cl-离子向氧化物晶格的渗透需要一定的时间,因此小孔腐蚀具有一个较长的诱导期。
从成相膜理论导出的机械破坏理论也能解释Cl-离子对钝化膜的破坏作用。由于Cl-离子的吸附,使膜/溶液界面的界面张力降低。当Cl-离子的吸附量增加到一定数量时,界面张力剧烈降低,以致在界面电荷的作用下发生了分离作用,即由于吸附阴离子的相互排斥,使原来牢固附着的氧化膜遭到破坏。Ilkovic在汞/溶液界面上曾做过实验,由于季胺阳离子的阴极吸附,在比零电荷电位负8V时,能使汞表面“爆炸”成一个带正电的汞溶胶。因此当一定数量的易变形的卤素阴离子(大于临界浓度时),在一个适当的正电位(大于击破电位)下足够强烈地吸附在氧化膜表面时,就能使固态的钝化膜发生破裂。这种机械破坏理论说明了氧化膜的破坏总是优先地发生在局部活性区域。
2)吸附理论的解释。由于溶液中的Cl-离子与溶解氧或OH-离子在Fe表面上的竞争吸附,在那些钝化膜较薄弱处原来吸附的氧被Cl-离子所置换,使吸附钝化膜遭到破坏,在此位置上的金属进入活性溶解状态。Uhlig认为吸附过程可用下式来表示:

式中,x为3~4,Me(Cl)xx-是具有高能量的过渡络合物,形成这种络合物的几率是很低的,但一经形成,络合物就脱离晶格进入溶液,使膜破坏。为了能达到上述平衡,就需要一定的Cl-离子浓度(临界浓度)。建立平衡后就有该反应的平衡电位(对应于孔蚀的击破电位),平衡建立所需的时间相当于小孔腐蚀的诱导期。
Cl-离子对Ti、Mo、Zr等金属的钝化膜的破坏作用很小,因此这些金属在高浓度氧化物溶液中仍能保持钝化状态。成膜理论认为这些金属的氯化物是不溶性的,因此在金属表面上生成了一层不溶性的氧化物保护膜。吸附理论认为,这些金属与氧的亲和力特别大,Cl-离子难以排挤掉金属表面上的吸附氧,因而能使钝化状态继续保持。
相关文章

在加载过程中,弹簧所吸收的能量称为变形能,以U 表示。对于自由放置的螺旋弹簧,在变形恢复时若没有与其他零件相摩擦,摩擦损失的能量等于零,则弹簧放出的能量和积蓄的能量相等。对于叠板弹簧、碟形弹簧及环形弹簧等,由于在过程中弹簧间变形相互摩擦,故有相当数量的摩擦功存在,致使卸载时的特性线低于加载时的特性线,如图3-35 所示。设计缓冲弹簧时,为保证其缓冲能力,应要求弹簧的变形能大于 被缓冲物体的动能。......
2025-09-29
图3.78不同力传导的三维说明对于应用中机械手最佳选择的问题并不容易回答。然而,当看到张角型机械手的夹持力变化过程与平动型机械手相比时,夹持力变化过程对于决策的相对重要性变得清晰。张角型机械手将根据手指的位置呈现不同的夹持力。相比之下,平动型机械手在整个手指行程中提供恒定的夹持力。......
2025-09-29

配体与金属原子或离子通过配位键形成的配合物叫金属配合物。金属配合物有诸多特殊的性质,如光、电、磁、催化、生物化学特性等,在科学实验和生产实践中应用广泛。通过配位化学和金属有机化学衍生得到的金属配合物在有机合成、有机催化等领域的作用也日益凸显。在众多的金属配合物中,二茂铁是一个典型的金属有机化合物。但对氧化的敏感性限制了它在合成中的应用,二茂铁的反应通常需要在隔绝空气的条件下进行。......
2025-09-29

如图10-6所示为PROFIBUS电缆,包括100%的铝箔屏蔽和65%的镀锡铜编织网屏蔽,具有最大的屏蔽效果。图10-11 总线连接器的终端电阻图10-12 终端电阻的具体设置5.通信处理器与PROFIBUS相关联的通信处理器主要包括:1)CP 342-5通信处理器;2)CP 342-5 FO通信处理器;3)CP 443-5通信处理器;4)用于PC/PG的通信处理器。......
2025-09-29
淀粉的链具有螺旋状结构,每6个葡萄糖基构成一螺旋圈,且羟基均指向内圈。直链淀粉遇碘呈蓝色,而支链淀粉遇碘呈紫红色,糊精与碘结合显色不同的主要原因是生成的淀粉-碘包合物不同。淀粉与碘生成包合物的颜色,与淀粉糖苷链的长度、聚合度或相对分子质量有关。直链淀粉的链长超过30个葡萄糖基,所以与碘作用呈现蓝色。根据淀粉与碘结合的特性,可以测定直链淀粉的含量。......
2025-09-29

常用的电镀修复技术有槽镀和电刷镀。槽镀时金属镀层种类繁多,设备维修中常用的有镀铬、镀铁、镀镍、镀铜及其合金等。图6.12电镀装置示意图电镀液由主盐、络合剂、附加盐、缓冲剂、阳极活化剂、添加剂等组成。镀铁不宜用于修复在高温、腐蚀环境、承受较大冲击载荷、干摩擦或磨料磨损条件下工作的零件。......
2025-09-29

例如:某照明灯的项目代号为“=S3+301-E3:2”,表示3号车间变电所301室3号照明灯的第2个端子。图1-33为某10kV线路过电流保护项目的项目代号、前缀及其分解图。图1-33 项目代号结构、前缀及其分解图1.高层代号对所给代号的项目而言,设备或系统中任何较高层次的代号都可称为高层代号。图1-34 位置代号说明示例图3.种类代号种类代号是用来识别项目种类的代号。端子代号是构成项目代号的一部分。......
2025-09-29
因为γ-Fe2O3在高于350℃时不稳定,所以高温氧化层的结构为α-Fe2O3/Fe3O4/FeO/Fe。在600℃以上时,FeO约占总氧化层量的90%以上。表1-5铁的各种氧化物的结构与性质Fe在570℃以上温度下氧化时生成的三层氧化物,其生长情况如图1-16所示。......
2025-09-29
相关推荐