实验类型 验证性教学时数 3操作视频一、实验目的加深对蛋白质胶体溶液稳定因素的认识。蛋白质变性并不一定沉淀,沉淀也并不一定变性。......
2023-11-04
实验十三 蛋白质—核酸相互作用分析
一、凝胶阻滞实验
凝胶阻滞实验又称为DNA迁移率变动实验,是在20世纪80年代初期出现的用于在体外研究DNA与蛋白质相互作用的一种特殊的凝胶电泳技术。该方法用来研究DNA与特异性蛋白的相互作用,通常是放射性标记的DNA片段与纯化蛋白、或提取物中的蛋白混合物相结合,然后在非变性凝胶中分析该产物。与游离DNA相比,蛋白-DNA复合物的迁移率将降低,因此,与游离DNA相对应,人们将观察到带中的“阻滞”。
该方法具有简单、快捷等优点,是分离纯化特定DNA结合蛋白质的一种典型的试验方法。该方法可用于检测DNA结合蛋白、RNA结合蛋白,并可通过加入特异性的抗体(supershift)来检测特定的蛋白质,并可进行未知蛋白的鉴定。
1.实验原理
在凝胶电泳中,由于电场的作用,裸露的DNA向正极移动的距离同其分子量的对数成反比,如果DNA分子上结合上一种蛋白质,那么由于分子量加大,其在凝胶中的迁移作用便会受到阻滞,朝正极移动的距离也就相应缩短了。当特定的DNA片段同细胞提取物混合之后,若其在凝胶电泳中的移动距离变小,说明它已同提取物中的某种蛋白质分子发生了结合作用。蛋白质可以与末端标记的核酸探针结合,这种复合物较无蛋白质结合的探针在非变性聚丙烯酰胺凝胶中的泳动速度要慢,即表现为相对滞后。而且根据复合物分子量的不同在凝胶中表现为不同的带型,每一条带即代表一种核酸结合蛋白。
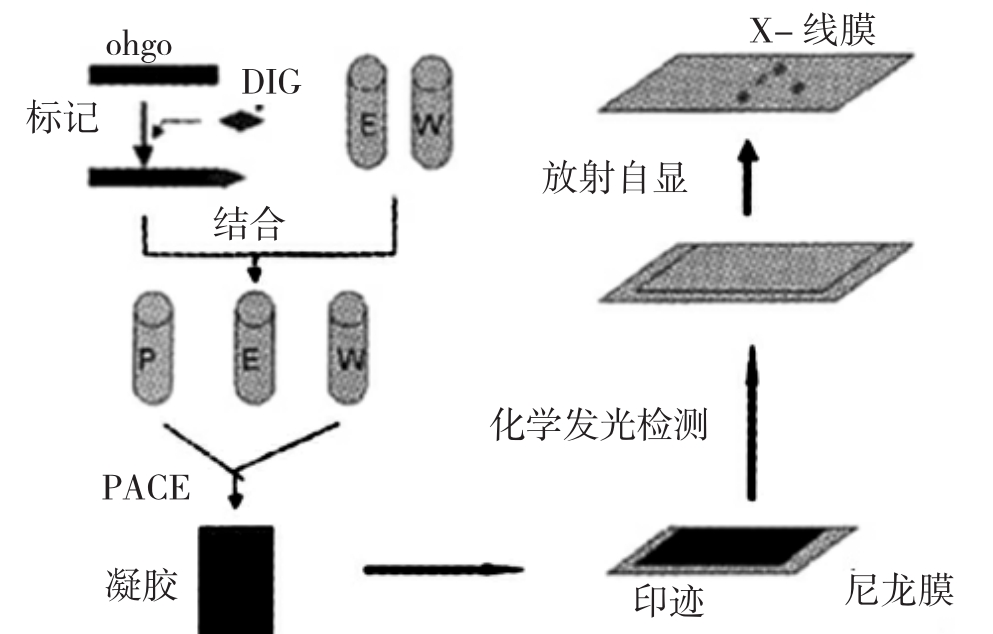
图3-11 凝胶阻滞实验原理
2.操作步骤
(1)对含有特异结合位点的DNA片段(亦称探针DNA)进行末端标记,如可用放射性同位素标记。
(2)与细胞蛋白质提取物共同孵育,进行DNA和蛋白质的结合反应。
(3)将上述共同孵育物进行非变性的聚丙烯酰胺凝胶电泳,并应用放射自显影技术显现具放射性标记的DNA条带位置。
(4)如果细胞蛋白质提取物中不存在可同放射性标记的探针DNA结合的蛋白质,那么所有放射性标记都将集中出现在凝胶的底部,反之,将会形成DNA-蛋白质复合物,由于凝胶阻滞的缘故,其特有的放射性标记的探针DNA条带将滞后出现在较靠近凝胶顶部的位置。
3.结果分析
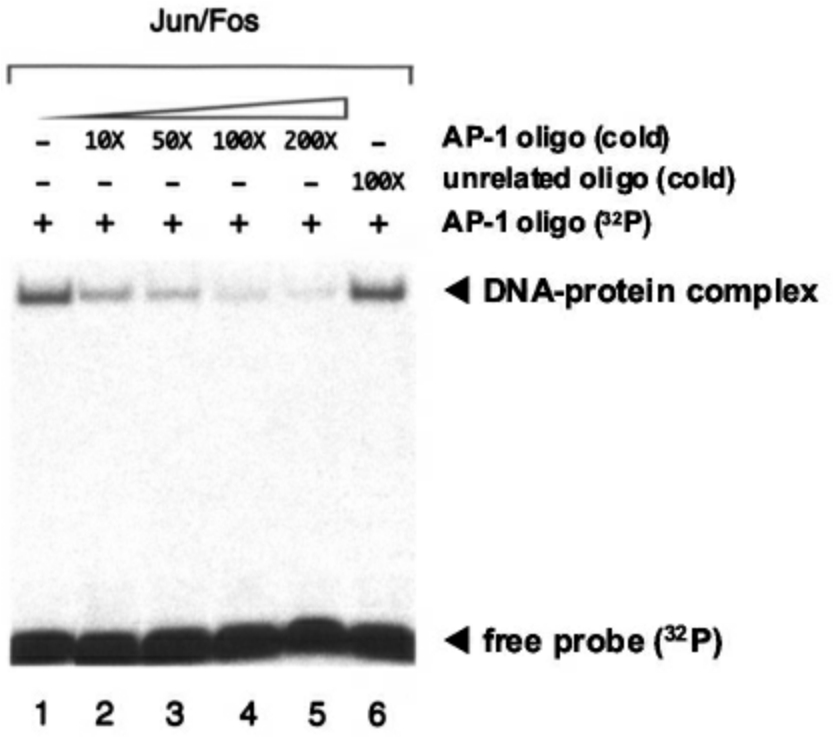
图3-12 凝胶滞后实验结果示意图
二、甲基化干扰实验
1.实验原理
利用硫酸二甲酯(DMS)使DNA分子中的嘌呤碱基G与A发生甲基化,被甲基化修饰的G和A,会干扰DNA结合蛋白与所在部位的DNA结合;甲基化的探针可被化学试剂哌啶特异地切割,从而得到不同分子量的DNA探针片段。而与蛋白质结合着的DNA位点则不会被切割。
用特殊方法将DNA从被修饰的碱基处进行切割,并经电泳分离,由于未结合蛋白的DNA可在随机修饰的位点被切割,而有结合蛋白的DNA在结合位点上未被修饰而不能被切割,由此可获得蛋白质与核酸定位结合的信息。
2.操作步骤
(1)将待测双链DNA片段中的一条单链的一端用放射性核素标记。
(2)甲基化修饰DNA探针,并使之与DNA结合蛋白反应。
(3)按电泳迁移率将游离DNA探针和DNA-蛋白质复合物分开,并回收。(www.chuimin.cn)
(4)分别用哌啶切割后在测序聚丙烯酰胺凝胶中电泳,放射自显影。
3.结果分析
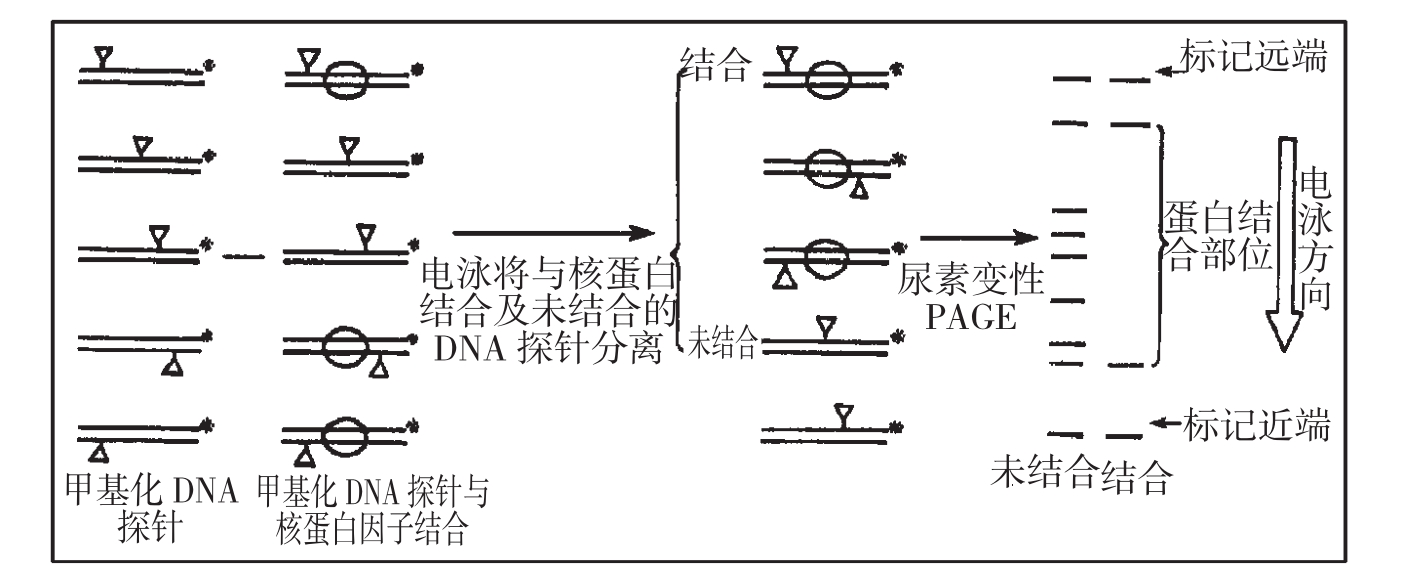
图3-13 DNA-蛋白质结合位点的检测结果图
三、DNaseI足迹分析
也称作DNaseI保护实验,是体外鉴定DNA结合蛋白在DNA分子上的结合位点的实验方法,是目前广泛用于蛋白质精确结合位点的研究方法。
1.实验原理
DNaseⅠ可以随机水解核苷酸中的磷酸二酯键,将DNA切成单核苷酸,而结合有蛋白质的DNA免于DNaseⅠ的水解。
将待测双链DNA片段中一条单链的一端选择性地进行末端标记,然后加入适当浓度的DNaseⅠ,使其在DNA链上随机形成缺口,经变性后电泳分离,放射自显影,即可形成以相差一个核苷酸为梯度的DNA条带。但当DNA片段与相应的序列特异性DNA结合蛋白结合后,DNA结合蛋白可保护相应的DNA序列不受DNaseⅠ的攻击,因而在放射自显影图谱上,DNA梯度条带在相应于DNA结合蛋白的结合区域中断,从而形成一空白区域,恰似蛋白质在DNA上留下的足迹,因而被形象地称作足迹法。如果同时进行DNA化学测序,即可判断出结合区的精确顺序。
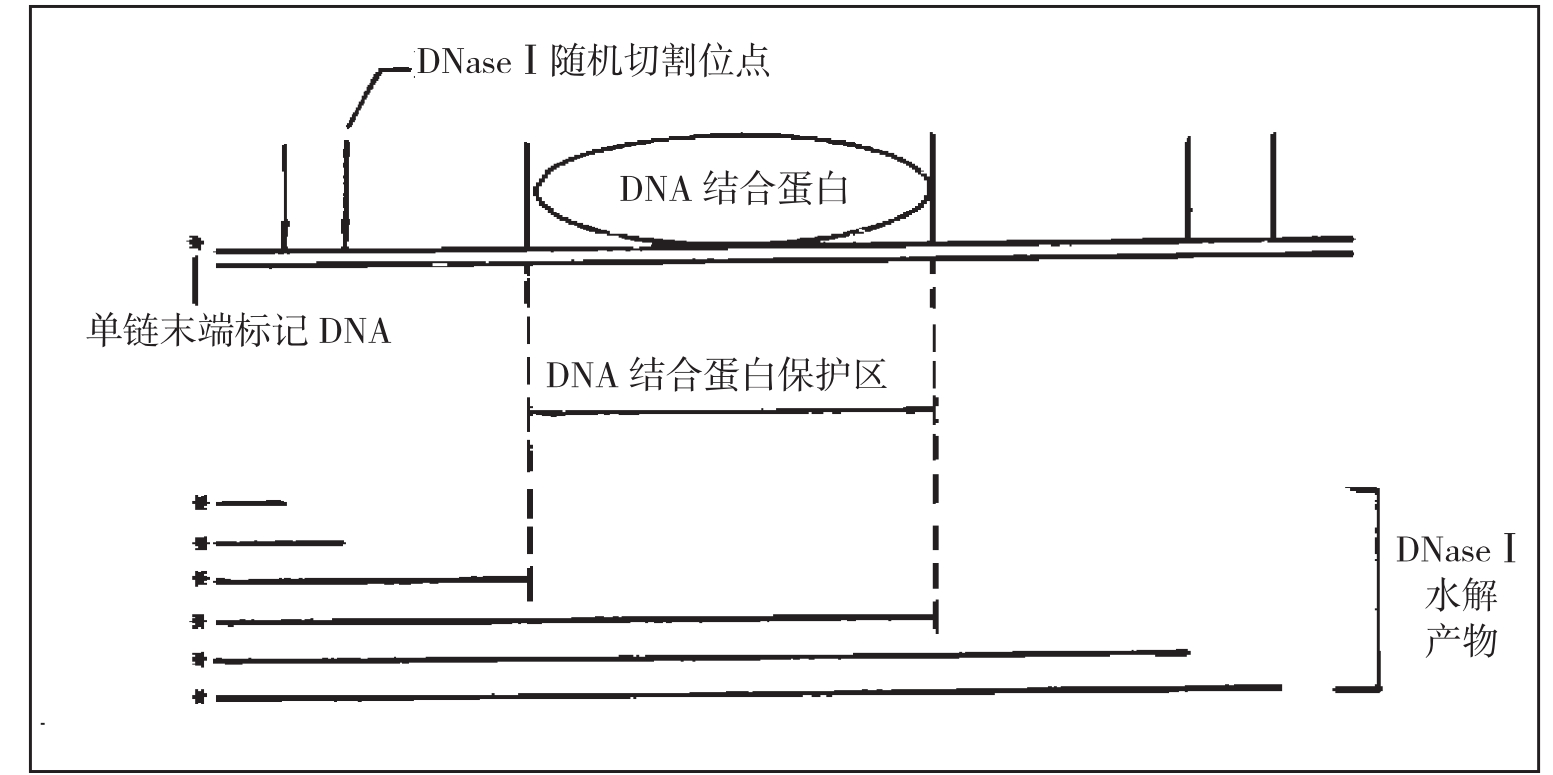
图3-14 DNaseⅠ足迹分析原理示意图
2.操作步骤(举例)
(1)探针制备及纯化
分别以BamHⅠ(标记Lowerstrand)和XhoⅠ(标记Upperstrand)酶切含104bp启动子片段的质粒pEGF-104,回收纯化后依次加入5μl10×Klenow缓冲液,2μl1.5mmol/L dGTP、dCTP和dTTP混合物,2μlα-32P-dATP,1μl的Klenow酶,加水补至50μl,混后,37℃温育20分钟,对两个3′黏端补平,并参入同位素进行标记。标记后用XhoⅠ和BamHⅠ进行酶切,电泳回收标记后的启动子片段备用。
(2)样品处理
本实验以LoVo细胞为A33基因表达的阳性细胞,293细胞为阴性对照。在Eppendorf管中混合下列成分:5μl5×结合缓冲液,一端标记的探针(cpm值为104),LoVo细胞核蛋白(分30μg和60μg两组)和293细胞核蛋白(60μg),poly(dT-dC)1μl,用去离子水调至50μl体积。不加核蛋白的对照组加入40μgBSA。室温放置30分钟。加入5μl10×Ca2+/Mg2+,室温放置1分钟,加入适量的DNaseⅠ室温作用1分钟,立即加入140μl终止液(0.2mol/LNaCl,20 mmol/LEDTApH8.0,1%SDS,0.25g/LcarrierRNA)终止反应,加入200μlTris饱和酚/氯仿(1∶1)抽提一次。将水相转移至另一Eppendorf管中,加入500μl无水乙醇于-80℃沉淀30分钟,12000g离心10分钟,弃上清。用75%乙醇漂洗2次,干燥后溶于3μl甲酰胺上样缓冲液中。
(3)G+A化学测序反应
在Eppendorf管中加入一端标记的探针(cpm值为105),0.5μg/μl的小牛胸腺DNA 2μl,用TE将总体积调整至10μl。加入1μl4%甲酸,37℃温育25分钟。将样品置于冰上冷却,加入150μl1mol/L哌啶,90℃温育30分钟。样品置于冰上5分钟,加入1ml正丁醇,震荡混匀,高速离心2分钟沉淀DNA,弃上清。用150μl1%SDS溶解沉淀,加入1ml正丁醇剧烈震荡,高速离心2分钟,去除上清。用0.5ml正丁醇漂洗2次,干燥后溶于10μl上样缓冲液中。
(4)电泳及结果分析
将足迹实验样品及G+A化学测序样品上6%聚丙烯酰胺变性凝胶电泳分离,50W恒功率电泳约1.5小时,至溴酚蓝前沿到达底端。将凝胶转移至滤纸上,80℃真空干燥1小时。于-80放射自显影48小时。
3.结果分析
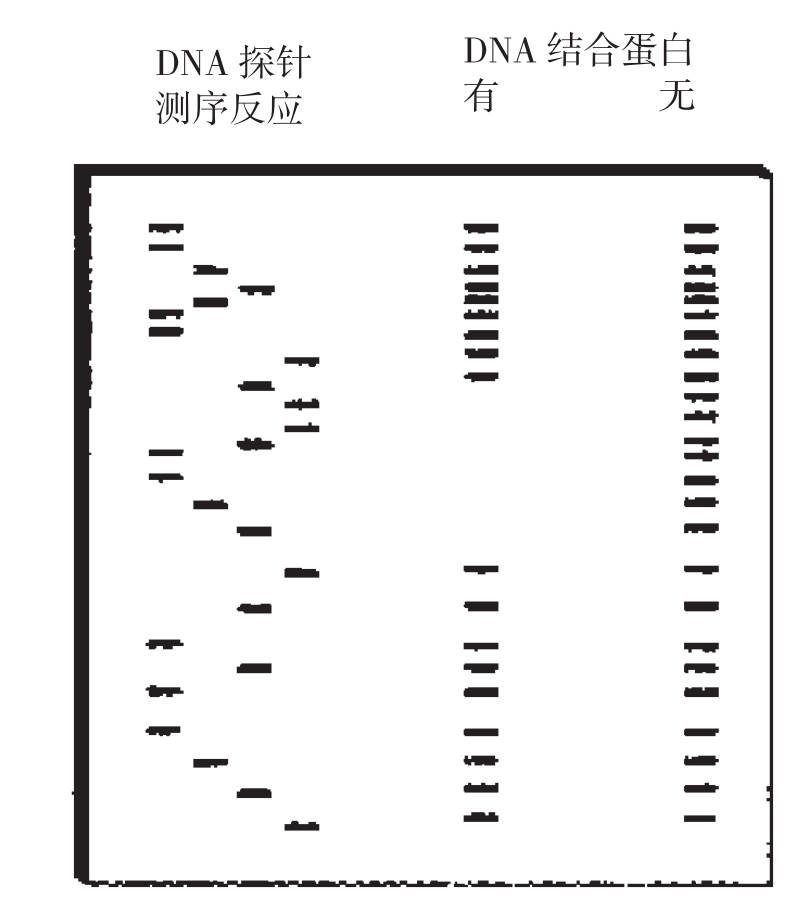
图3-15 DNA-蛋白质结合位点的检测结果示意图
有关生物技术制药实验的文章

课题设计细胞内的核酸包括DNA与RNA两种分子,均与蛋白质结合成核蛋白。DNA与RNA性质上的差异决定了两者的最适分离与纯化的条件是不一样的。临床常见的标本有血液、尿液、唾液、组织及培养细胞等;核酸分离与纯化的方法非常多,如何恰当地收集与准备材料,选择适宜的分离与纯化方法是一个首要的问题。近年来,有关试剂盒的开发与自动化仪器的使用,能批量制备核酸样品,大大提高了分离与纯化的效率。......
2023-11-04

之后计算求得该组5个制品减重百分数的平均值xi,作为该组实验的结果。表6-9 实验结果表6-10 实验结果表6-11 实验结果表6-9~表6-11中,每一行为一组实验数据,各因素下为对应的水平号。表6-10~表6-11中实验号5、13、14的实验结果明显小于其他数据,后续数据处理中保留了这些数据,没有作为奇异项处理。图6-32 一个注塑周期的实验数据图所有数据统一在一个时间轴下记录,为后续数据分析提供了便利。......
2023-07-02

实验类型 验证性教学时数 3一、实验目的学习紫外吸收法测定核酸含量的原理。因而可以核酸的紫外吸收性进行核酸的定量测定。当核酸变性降解时,其紫外吸收强度显著增加,称为增色效应;反之,变性DNA复性后,吸光度降低,称为减色效应。若待测的核酸制品中混有大量的具有紫外吸收的杂质,则测定误差较大,应设法除去。不纯的样品不能用紫外吸收值作定量测定。......
2023-11-04

可用于蛋白质的定性或定量测定。因此,一切蛋白质或二肽以上的多肽都有双缩脲反应,但有双缩脲反应的物质不一定都是蛋白质或多肽。因此,虽然蛋白质和氨基酸均有茚三酮反应,但能与茚三酮呈阳性反应的不一定就是蛋白质或氨基酸。含有色氨酸的蛋白质也有此反应。......
2023-11-06
实验十四蛋白质印迹分析1.实验目的掌握Westernblot技术的基本原理,学习Western blot技术的实验方法,了解Westernblot技术的应用范围。......
2023-12-07
实验十二二维双向凝胶电泳分析鉴定蛋白质双向凝胶电泳是目前蛋白质组研究中最有效的分析鉴定技术之一。第二向SDS-PAGE有垂直板电泳和水平超薄胶电泳两种做法,可分离10~100kD分子量的蛋白质。......
2023-12-07
核酸及其衍生物核苷酸、核苷、嘌呤和嘧啶具有吸收紫外光的性质,其吸收高峰在260nm波长处。因此,测定未知浓度核酸溶液的A260值,即可计算出其中RNA或DNA的含量。通常蛋白质的最大吸收峰在280nm波长处,在260nm处的吸收值仅为核酸的1/10或更低,因此对于含有微量蛋白质的核酸样品,测定误差较小。选择厚度为1cm的石英比色杯,于紫外分光光度计上260nm波长处测定A值。......
2023-11-06
相关推荐